Brand Name
Pamidronate
View Brand InformationFDA approval date: July 28, 2003
Classification: Bisphosphonate
Form: Injection
What is Pamidronate?
Hypercalcemia of Malignancy Pamidronate disodium, in conjunction with adequate hydration, is indicated for the treatment of moderate or severe hypercalcemia associated with malignancy, with or without bone metastases. Patients who have either epidermoid or non-epidermoid tumors respond to treatment with pamidronate disodium. Vigorous saline hydration, an integral part of hypercalcemia therapy, should be initiated promptly and an attempt should be made to restore the urine output to about 2 L/day throughout treatment. Mild or asymptomatic hypercalcemia may be treated with conservative measures . Patients should be hydrated adequately throughout the treatment, but overhydration, especially in those patients who have cardiac failure, must be avoided. Diuretic therapy should not be employed prior to correction of hypovolemia. The safety and efficacy of pamidronate disodium in the treatment of hypercalcemia associated with hyperparathyroidism or with other non-tumor-related conditions has not been established. Paget’s Disease Pamidronate disodium is indicated for the treatment of patients with moderate to severe Paget’s disease of bone. The effectiveness of pamidronate disodium was demonstrated primarily in patients with serum alkaline phosphatase ≥ 3 times the upper limit of normal. Pamidronate disodium therapy in patients with Paget’s disease has been effective in reducing serum alkaline phosphatase and urinary hydroxyproline levels by ≥ 50% in at least 50% of patients, and by ≥ 30% in at least 80% of patients. Pamidronate disodium therapy has also been effective in reducing these biochemical markers in patients with Paget’s disease who failed to respond, or no longer responded to other treatments. Osteolytic Bone Metastases of Breast Cancer and Osteolytic Lesions of Multiple Myeloma Pamidronate disodium is indicated, in conjunction with standard antineoplastic therapy, for the treatment of osteolytic bone metastases of breast cancer and osteolytic lesions of multiple myeloma. The pamidronate disodium treatment effect appeared to be smaller in the study of breast cancer patients receiving hormonal therapy than in the study of those receiving chemotherapy, however, overall evidence of clinical benefit has been demonstrated.
Approved To Treat
Top Global Experts



Save this treatment for later
Not sure about your diagnosis?
Tired of the same old research?
Related Clinical Trials
CHronic Nonbacterial Osteomyelitis International Registry (CHOIR)
Summary: The objective of the study is to establish a prospective disease registry for chronic recurrent multifocal osteomyelitis (CRMO)/chronic nonbacterial osteomyelitis (CNO) in order to investigate the natural history of the disease and the responses of patients to different clinical managements over 10 years.
Related Latest Advances
Brand Information
Pamidronate Disodium (Pamidronate Disodium)
1DESCRIPTION
Pamidronate Disodium is a sterile bone-resorption inhibitor available in 30 mg and 90 mg vials for intravenous administration. The pamidronate disodium obtained by combining pamidronic acid and sodium hydroxide is provided in a sterile, ready to use solution for injection. Each mL of the 30 mg vial contains, 3 mg Pamidronate Disodium, 47 mg Mannitol, USP; Water for Injection, USP, q.s.; Phosphoric acid to adjust pH. Each mL of the 90 mg vial contains, 9 mg Pamidronate Disodium, 37.5 mg Mannitol, USP; Water for Injection, USP, q.s.; Phosphoric acid to adjust pH. The pH of a 1% solution of pamidronate disodium in distilled water is approximately 8.3. Pamidronate, a member of the group of chemical compounds known as bisphosphonates, is an analog of pyrophosphate. Pamidronate disodium is designated chemically as phosphonic acid (3-amino-1-hydroxypropylidene) bis-, disodium salt, and its structural formula is:
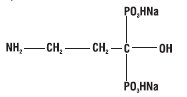
Pamidronate disodium is soluble in water and in 2N sodium hydroxide, sparingly soluble in 0.1N hydrochloric acid and in 0.1N acetic acid, and practically insoluble in organic solvents. Its molecular formula is C
Inactive Ingredients: Mannitol, Phosphoric acid (for adjustment to pH range of 6.0 to 7.0) and Water for Injection.
2CLINICAL PHARMACOLOGY
The principal pharmacologic action of pamidronate disodium is inhibition of bone resorption. Although the mechanism of antiresorptive action is not completely understood, several factors are thought to contribute to this action. Pamidronate disodium adsorbs to calcium phosphate (hydroxyapatite) crystals in bone and may directly block dissolution of this mineral component of bone.
2.1Pharmacokinetics
Cancer patients (n=24) who had minimal or no bony involvement were given an intravenous infusion of 30, 60, or 90 mg of pamidronate disodium over 4 hours and 90 mg of pamidronate disodium over 24 hours (
2.2Distribution
The mean ± SD body retention of pamidronate was calculated to be 54 ± 16% of the dose over 120 hours.
2.3Metabolism
Pamidronate is not metabolized and is exclusively eliminated by renal excretion.
2.4Excretion
After administration of 30, 60, and 90 mg of pamidronate disodium over 4 hours, and 90 mg of pamidronate disodium over 24 hours, an overall mean ± SD of 46 ± 16% of the drug was excreted unchanged in the urine within 120 hours. Cumulative urinary excretion was linearly related to dose. The mean ± SD elimination half-life is 28 ± 7 hours. Mean ± SD total and renal clearances of pamidronate were 107 ± 50 mL/min and 49 ± 28 mL/min, respectively. The rate of elimination from bone has not been determined.
2.5Special Populations
There are no data available on the effects of age, gender, or race on the pharmacokinetics of pamidronate.
2.5.1Pediatric
Pamidronate is not labeled for use in the pediatric population.
2.5.2Renal Insufficiency
The pharmacokinetics of pamidronate were studied in cancer patients (n=19) with normal and varying degrees of renal impairment. Each patient received a single 90 mg dose of pamidronate disodium infused over 4 hours. The renal clearance of pamidronate in patients was found to closely correlate with creatinine clearance (see
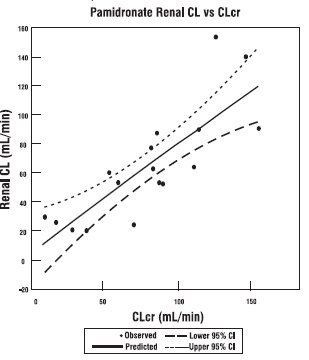
The lines are the mean prediction line and 95% confidence intervals.
2.5.3Hepatic Insufficiency
The pharmacokinetics of pamidronate were studied in male cancer patients at risk for bone metastases with normal hepatic function (n=6) and mild to moderate hepatic dysfunction (n=7). Each patient received a single 90 mg dose of pamidronate disodium infused over 4 hours. Although there was a statistically significant difference in the pharmacokinetics between patients with normal and impaired hepatic function, the difference was not considered clinically relevant. Patients with hepatic impairment exhibited higher mean AUC (53%) and C
2.5.4Drug – Drug Interactions
There are no human pharmacokinetic data for drug interactions with pamidronate disodium.
After intravenous administration of radiolabeled pamidronate in rats, approximately 50% to 60% of the compound was rapidly adsorbed by bone and slowly eliminated from the body by the kidneys. In rats given 10 mg/kg bolus injections of radiolabeled pamidronate disodium, approximately 30% of the compound was found in the liver shortly after administration and was then redistributed to bone or eliminated by the kidneys over 24 to 48 hours. Studies in rats injected with radiolabeled pamidronate disodium showed that the compound was rapidly cleared from the circulation and taken up mainly by bones, liver, spleen, teeth, and tracheal cartilage. Radioactivity was eliminated from most soft tissues within 1 to 4 days; was detectable in liver and spleen for 1 and 3 months, respectively; and remained high in bones, trachea, and teeth for 6 months after dosing. Bone uptake occurred preferentially in areas of high bone turnover. The terminal phase of elimination half-life in bone was estimated to be approximately 300 days.
2.6Pharmacodynamics
Serum phosphate levels have been noted to decrease after administration of pamidronate disodium, presumably because of decreased release of phosphate from bone and increased renal excretion as parathyroid hormone levels, which are usually suppressed in hypercalcemia associated with malignancy, return toward normal. Phosphate therapy was administered in 30% of the patients in response to a decrease in serum phosphate levels. Phosphate levels usually returned toward normal within 7 to 10 days.
Urinary calcium/creatinine and urinary hydroxyproline/creatinine ratios decrease and usually return to within or below normal after treatment with pamidronate disodium. These changes occur within the first week after treatment, as do decreases in serum calcium levels, and are consistent with an antiresorptive pharmacologic action.
2.7Hypercalcemia of Malignancy
Osteoclastic hyperactivity resulting in excessive bone resorption is the underlying pathophysiologic derangement in metastatic bone disease and hypercalcemia of malignancy. Excessive release of calcium into the blood as bone is resorbed results in polyuria and gastrointestinal disturbances, with progressive dehydration and decreasing glomerular filtration rate. This, in turn, results in increased renal resorption of calcium, setting up a cycle of worsening systemic hypercalcemia. Correction of excessive bone resorption and adequate fluid administration to correct volume deficits are therefore essential to the management of hypercalcemia.
Most cases of hypercalcemia associated with malignancy occur in patients who have breast cancer; squamous-cell tumors of the lung or head and neck; renal-cell carcinoma; and certain hematologic malignancies, such as multiple myeloma and some types of lymphomas. A few less-common malignancies, including vasoactive intestinal-peptide-producing tumors and cholangiocarcinoma, have a high incidence of hypercalcemia as a metabolic complication. Patients who have hypercalcemia of malignancy can generally be divided into two groups, according to the pathophysiologic mechanism involved.
In humoral hypercalcemia, osteoclasts are activated and bone resorption is stimulated by factors such as parathyroid-hormone-related protein, which are elaborated by the tumor and circulate systemically. Humoral hypercalcemia usually occurs in squamous-cell malignancies of the lung or head and neck or in genitourinary tumors such as renal-cell carcinoma or ovarian cancer. Skeletal metastases may be absent or minimal in these patients.
Extensive invasion of bone by tumor cells can also result in hypercalcemia due to local tumor products that stimulate bone resorption by osteoclasts. Tumors commonly associated with locally mediated hypercalcemia include breast cancer and multiple myeloma.
Total serum calcium levels in patients who have hypercalcemia of malignancy may not reflect the severity of hypercalcemia, since concomitant hypoalbuminemia is commonly present. Ideally, ionized calcium levels should be used to diagnose and follow hypercalcemic conditions; however, these are not commonly or rapidly available in many clinical situations. Therefore, adjustment of the total serum calcium value for differences in albumin levels is often used in place of measurement of ionized calcium; several nomograms are in use for this type of calculation (see
2.7.1Clinical Trials
In one double-blind clinical trial, 52 patients who had hypercalcemia of malignancy were enrolled to receive 30 mg, 60 mg, or 90 mg of pamidronate disodium as a single 24 hour intravenous infusion if their corrected serum calcium levels were ≥12 mg/dL after 48 hours of saline hydration.
The mean baseline-corrected serum calcium for the 30 mg, 60 mg, and 90 mg groups were 13.8 mg/dL, 13.8 mg/dL, and 13.3 mg/dL, respectively.
The majority of patients (64%) had decreases in albumin-corrected serum calcium levels by 24 hours after initiation of treatment. Mean-corrected serum calcium levels at days 2 to 7 after initiation of treatment with pamidronate disodium were significantly reduced from baseline in all three dosage groups. As a result, by 7 days after initiation of treatment with pamidronate disodium, 40%, 61%, and 100% of the patients receiving 30 mg, 60 mg, and 90 mg of pamidronate disodium, respectively, had normal-corrected serum calcium levels. Many patients (33% to 53%) in the 60 mg and 90 mg dosage groups continued to have normal-corrected serum calcium levels, or a partial response (≥15% decrease of corrected serum calcium from baseline), at Day 14.
In a second double-blind, controlled clinical trial, 65 cancer patients who had corrected serum calcium levels of ≥12.0 mg/dL after at least 24 hours of saline hydration were randomized to receive either 60 mg of pamidronate disodium as a single 24 hour intravenous infusion or 7.5 mg/kg of etidronate disodium as a 2 hour intravenous infusion daily for 3 days. Thirty patients were randomized to receive pamidronate disodium and 35 to receive etidronate disodium.
The mean baseline-corrected serum calcium for the pamidronate disodium 60 mg and etidronate disodium groups were 14.6 mg/dL and 13.8 mg/dL, respectively.
By Day 7, 70% of the patients in the pamidronate disodium group and 41% of the patients in the etidronate disodium group had normal-corrected serum calcium levels (P<0.05). When partial responders (≥15% decrease of serum calcium from baseline) were also included, the response rates were 97% for the pamidronate disodium group and 65% for the etidronate disodium group (P<0.01). Mean-corrected serum calcium for the pamidronate disodium and etidronate disodium groups decreased from baseline values to 10.4 and 11.2 mg/dL, respectively, on Day 7. At Day 14, 43% of patients in the pamidronate disodium group and 18% of patients in the etidronate disodium group still had normal-corrected serum calcium levels, or maintenance of a partial response. For responders in the pamidronate disodium and etidronate disodium groups, the median duration of response was similar (7 and 5 days, respectively). The time course of effect on corrected serum calcium is summarized in the following table.
In a third multicenter, randomized, parallel double-blind trial, a group of 69 cancer patients with hypercalcemia was enrolled to receive 60 mg of pamidronate disodium as a 4 or 24 hour infusion, which was compared to a saline treatment group. Patients who had a corrected serum calcium level of ≥12.0 mg/dL after 24 hours of saline hydration were eligible for this trial.
The mean baseline-corrected serum calcium levels for pamidronate disodium 60 mg 4 hour infusion, pamidronate disodium 60 mg 24 hour infusion, and saline infusion were 14.2 mg/dL, 13.7 mg/dL, and 13.7 mg/dL, respectively.
By Day 7 after initiation of treatment, 78%, 61%, and 22% of the patients had normal-corrected serum calcium levels for the 60 mg 4 hour infusion, 60 mg 24 hour infusion, and saline infusion, respectively. At Day 14, 39% of the patients in the pamidronate disodium 60 mg 4 hour infusion group and 26% of the patients in the pamidronate disodium 60 mg 24 hour infusion group had normal-corrected serum calcium levels or maintenance of a partial response.
For responders, the median duration of complete responses was 4 days and 6.5 days for pamidronate disodium 60 mg 4 hour infusion and pamidronate disodium 60 mg 24 hour infusion, respectively.
In all three trials, patients treated with pamidronate disodium had similar response rates in the presence or absence of bone metastases. Concomitant administration of furosemide did not affect response rates.
Thirty-two patients who had recurrent or refractory hypercalcemia of malignancy were given a second course of 60 mg of pamidronate disodium over a 4 or 24 hour period. Of these, 41% showed a complete response and 16% showed a partial response to the retreatment, and these responders had about a 3 mg/dL fall in mean-corrected serum calcium levels 7 days after retreatment.
In a fourth multicenter, randomized, double-blind trial, 103 patients with cancer and hypercalcemia (corrected serum calcium ≥12.0 mg/dL) received 90 mg of pamidronate disodium as a 2 hour infusion. The mean baseline corrected serum calcium was 14.0 mg/dL. Patients were not required to receive IV hydration prior to drug administration, but all subjects did receive at least 500 mL of IV saline hydration concomitantly with the pamidronate infusion. By Day 10 after drug infusion, 70% of patients had normal corrected serum calcium levels (<10.8 mg/dL).
2.8Paget’s Disease
Paget’s disease of bone (osteitis deformans) is an idiopathic disease characterized by chronic, focal areas of bone destruction complicated by concurrent excessive bone repair, affecting one or more bones. These changes result in thickened but weakened bones that may fracture or bend under stress. Signs and symptoms may be bone pain, deformity, fractures, neurological disorders resulting from cranial and spinal nerve entrapment and from spinal cord and brain stem compression, increased cardiac output to the involved bone, increased serum alkaline phosphatase levels (reflecting increased bone formation) and/or urine hydroxyproline excretion (reflecting increased bone resorption).
2.8.1Clinical Trials
In one double-blind clinical trial, 64 patients with moderate to severe Paget’s disease of bone were enrolled to receive 5 mg, 15 mg, or 30 mg of pamidronate disodium as a single 4 hour infusion on 3 consecutive days, for total doses of 15 mg, 45 mg, and 90 mg of pamidronate disodium.
The mean baseline serum alkaline phosphatase levels were 1409 U/L, 983 U/L, and 1085 U/L, and the mean baseline urine hydroxyproline/creatinine ratios were 0.25, 0.19, and 0.19 for the 15 mg, 45 mg, and 90 mg groups, respectively.
The effects of pamidronate disodium on serum alkaline phosphatase (SAP) and urine hydroxyproline/creatinine ratios (UOHP/C) are summarized in the following table:
The median maximum percent decreases from baseline in serum alkaline phosphatase and urine hydroxyproline/creatinine ratios were 25%, 41%, and 57%, and 25%, 47%, and 61% for the 15 mg, 45 mg, and 90 mg groups, respectively. The median time to response (≥50% decrease) for serum alkaline phosphatase was approximately 1 month for the 90 mg group, and the response duration ranged from 1 to 372 days.
No statistically significant differences between treatment groups, or statistically significant changes from baseline were observed for the bone pain response, mobility, and global evaluation in the 45 mg and 90 mg groups. Improvement in radiologic lesions occurred in some patients in the 90 mg group.
Twenty-five patients who had Paget’s disease were retreated with 90 mg of pamidronate disodium. Of these, 44% had a ≥50% decrease in serum alkaline phosphatase from baseline after treatment, and 39% had a ≥50% decrease in urine hydroxyproline/creatinine ratio from baseline after treatment.
2.9Osteolytic Bone Metastases of Breast Cancer and Osteolytic Lesions of Multiple Myeloma
Osteolytic bone metastases commonly occur in patients with multiple myeloma or breast cancer. These cancers demonstrate a phenomenon known as osteotropism, meaning they possess an extraordinary affinity for bone. The distribution of osteolytic bone metastases in these cancers is predominantly in the axial skeleton, particularly the spine, pelvis, and ribs, rather than the appendicular skeleton, although lesions in the proximal femur and humerus are not uncommon. This distribution is similar to the red bone marrow in which slow blood flow possibly assists attachment of metastatic cells. The surface-to-volume ratio of trabecular bone is much higher than cortical bone, and therefore disease processes tend to occur more floridly in trabecular bone than at sites of cortical tissue.
These bone changes can result in patients having evidence of osteolytic skeletal destruction leading to severe bone pain that requires either radiation therapy or narcotic analgesics (or both) for symptomatic relief. These changes also cause pathologic fractures of bone in both the axial and appendicular skeleton. Axial skeletal fractures of the vertebral bodies may lead to spinal cord compression or vertebral body collapse with significant neurologic complications. Also, patients may experience episode(s) of hypercalcemia.
2.9.1Clinical Trials
In a double-blind, randomized, placebo-controlled trial, 392 patients with advanced multiple myeloma were enrolled to receive pamidronate disodium or placebo in addition to their underlying antimyeloma therapy to determine the effect of pamidronate disodium on the occurrence of skeletal-related events (SREs). SREs were defined as episodes of pathologic fractures, radiation therapy to bone, surgery to bone, and spinal cord compression. Patients received either 90 mg of pamidronate disodium or placebo as a monthly 4 hour intravenous infusion for 9 months. Of the 392 patients, 377 were evaluable for efficacy (196 pamidronate disodium, 181 placebo). The proportion of patients developing any SRE was significantly smaller in the pamidronate disodium group (24% vs 41%, P<0.001), and the mean skeletal morbidity rate (#SRE/year) was significantly smaller for pamidronate disodium patients than for placebo patients (mean: 1.1 vs 2.1, P<0.02). The times to the first SRE occurrence, pathologic fracture, and radiation to bone were significantly longer in the pamidronate disodium group (P=.001, .006, and .046, respectively). Moreover, fewer pamidronate disodium patients suffered any pathologic fracture (17% vs 30%, P=.004) or needed radiation to bone (14% vs 22%, P=.049).
In addition, decreases in pain scores from baseline occurred at the last measurement for those pamidronate disodium patients with pain at baseline (P=.026) but not in the placebo group. At the last measurement, a worsening from baseline was observed in the placebo group for the Spitzer quality of life variable (P<.001) and ECOG performance status (P<.011) while there was no significant deterioration from baseline in these parameters observed in pamidronate disodium-treated patients.*
After 21 months, the proportion of patients experiencing any skeletal event remained significantly smaller in the pamidronate disodium group than the placebo group (P=.015). In addition, the mean skeletal morbidity rate (#SRE/year) was 1.3 vs 2.2 for pamidronate disodium patients vs placebo patients (P=.008), and time to first SRE was significantly longer in the pamidronate disodium group compared to placebo (P=.016). Fewer pamidronate disodium patients suffered vertebral pathologic fractures (16% vs 27%, P=.005). Survival of all patients was not different between treatment groups.
Two double-blind, randomized, placebo-controlled trials compared the safety and efficacy of 90 mg of pamidronate disodium infused over 2 hours every 3 to 4 weeks for 24 months to that of placebo in preventing SREs in breast cancer patients with osteolytic bone metastases who had one or more predominantly lytic metastases of at least 1 cm in diameter: one in patients being treated with antineoplastic chemotherapy and the second in patients being treated with hormonal antineoplastic therapy at trial entry.
382 patients receiving chemotherapy were randomized, 185 to pamidronate disodium and 197 to placebo. 372 patients receiving hormonal therapy were randomized, 182 to pamidronate disodium and 190 to placebo. All but three patients were evaluable for efficacy. Patients were followed for 24 months of therapy or until they went off study. Median duration of follow-up was 13 months in patients receiving chemotherapy and 17 months in patients receiving hormone therapy. Twenty-five percent of the patients in the chemotherapy study and 37% of the patients in the hormone therapy study received pamidronate disodium for 24 months. The efficacy results are shown in the table below:
Bone lesion response was radiographically assessed at baseline and at 3, 6, and 12 months. The complete + partial response rate was 33% in pamidronate disodium patients and 18% in placebo patients treated with chemotherapy (P=.001). No difference was seen between pamidronate disodium and placebo in hormonally-treated patients.
Pain and analgesic scores, ECOG performance status and Spitzer quality of life index were measured at baseline and periodically during the trials. The changes from baseline to the last measurement carried forward are shown in the following table:
3CONTRAINDICATIONS
Pamidronate disodium is contraindicated in patients with clinically significant hypersensitivity to pamidronate disodium or other bisphosphonates.
4OVERDOSAGE
There have been several cases of drug maladministration of intravenous pamidronate disodium in hypercalcemia patients with total doses of 225 mg to 300 mg given over 2
In addition, one obese woman (95 kg) who was treated with 285 mg of pamidronate disodium/day for 3 days experienced high fever (39.5°C), hypotension (from 170/90 mmHg to 90/60 mmHg), and transient taste perversion, noted about 6 hours after the first infusion. The fever and hypotension were rapidly corrected with steroids.
If overdosage occurs, symptomatic hypocalcemia could also result; such patients should be treated with short-term intravenous calcium.
5HOW SUPPLIED
Pamidronate Disodium Injection is available as follows:
Vials – 3 mg/mL, 10 mL vial - each contains 30 mg of Pamidronate Disodium and 470 mg of Mannitol, USP in 10 mL Water for Injection, USP.
Carton of 1 vial. NDC 67457-430-10.
Vials – 9 mg/mL, 10 mL vial - each contains 90 mg of Pamidronate Disodium and 375 mg of Mannitol, USP in 10 mL Water for Injection, USP.
Carton of 1 vial. NDC 67457-446-10.
Storage: Store at 20° to 25°C (68°to 77°F.) [See USP Controlled Room Temperature.]
Manufactured for:
Manufactured by:
1033247
1200005570
July 2022
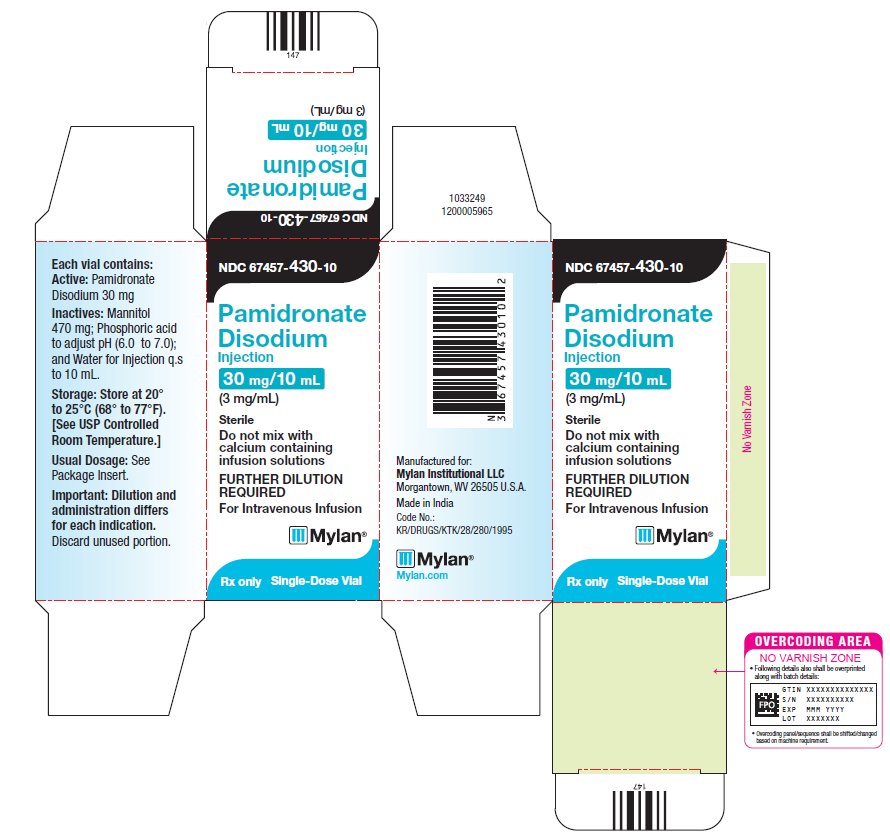
6PRINCIPAL DISPLAY PANEL – 3 mg/mL
NDC 67457-430-10
Pamidronate
30 mg/10 mL
Sterile
Do not mix with
FURTHER DILUTION
For Intravenous Infusion
Mylan
Rx only
Single-Dose Vial
Each vial contains:
Active: Pamidronate
Disodium 30 mg
Active: Pamidronate
Disodium 30 mg
Inactives: Mannitol
Storage: Store at 20°
Usual Dosage: See
Package Insert.
Package Insert.
Important: Dilution and
Discard unused portion.
Manufactured for:
Made in India
Code No.:
KR/DRUGS/KTK/28/280/1995
Mylan.com
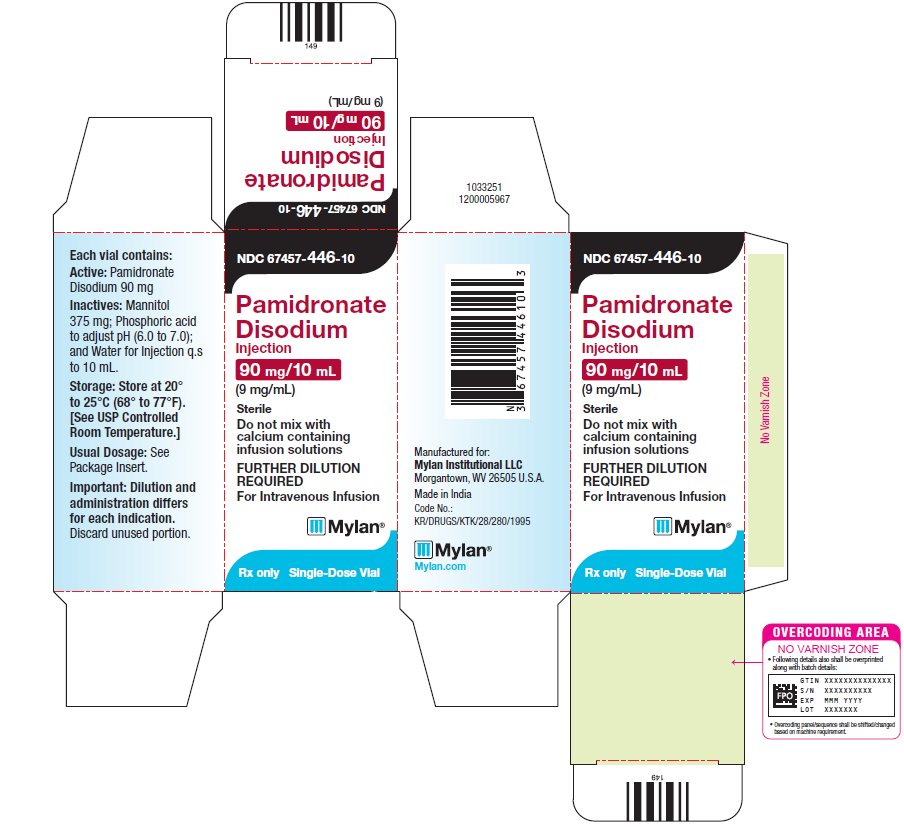
7PRINCIPAL DISPLAY PANEL – 9 mg/mL
NDC 67457-446-10
Pamidronate
90 mg/10 mL
Sterile
Do not mix with
FURTHER DILUTION
For Intravenous Infusion
Mylan
Rx only
Single-Dose Vial
Each vial contains:
Active: Pamidronate
Disodium 90 mg
Active: Pamidronate
Disodium 90 mg
Inactives: Mannitol
375 mg; Phosphoric acid
to adjust pH (6.0 to 7.0);
and Water for Injection q.s
to 10 mL.
375 mg; Phosphoric acid
to adjust pH (6.0 to 7.0);
and Water for Injection q.s
to 10 mL.
Storage: Store at 20°
Usual Dosage: See
Package Insert.
Package Insert.
Important: Dilution and
Discard unused portion.
Manufactured for:
Made in India
Code No.:
KR/DRUGS/KTK/28/280/1995
Mylan.com