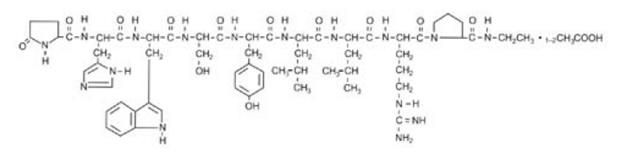
Leuprolide Acetate
What is Lupron (Leuprolide Acetate)?
Approved To Treat

Related Clinical Trials
Summary: This phase II trial studies the effects of stereotactic body radiation therapy (SBRT) and the timing of treatment with androgen receptor pathway inhibitor (ARPI) plus androgen deprivation therapy (ADT) in treating patients with hormone sensitive prostate cancer that has spread from where it first started to other places in the body (metastatic), and that has come back after a period of improvement...
Summary: This research study is studying a combination of HER2-directed therapies (trastuzumab and pertuzumab) and hormonal therapy as a treatment after surgery for hormone receptor positive breast cancer. The study drugs involved in this study are: * A combination of trastuzumab and pertuzumab given as an injection under the skin (PHESGO) * Hormonal (endocrine) Treatment
Summary: To verify the non-inferiority of KLH-2109 to leuprorelin acetate in a double-blind manner in terms of efficacy in endometriosis patients with pelvic pain.
Related Latest Advances
Brand Information
- Hypersensitivity to gonadotropin-releasing hormone (GnRH), GnRH agonist analogs, including leuprolide acetate, or any of the excipients in LUPRON DEPOT 3.75 mg
- Undiagnosed abnormal uterine bleeding
- Pregnancy
- Loss of Bone Mineral Density
- Hypersensitivity Reactions
- Initial Flare of Symptoms with Management of Endometriosis
- Convulsions
- Clinical Depression
- Body as a whole: Hypersensitivity reactions including anaphylaxis, localized reactions including induration and abscess at the site of injection
- Nervous/Psychiatric System: Mood swings, including depression; suicidal ideation and attempt; convulsion, peripheral neuropathy, paralysis
- Hepato-biliary system: Serious liver injury
- Skin reactions: erythema multiforme, dermatitis bullous, dermatitis exfoliative, Stevens-Johnson Syndrome (SJS) and Toxic Epidermal Necrolysis (TEN)
- Injury, poisoning and procedural complications: Spinal fracture
- Investigations: Decreased white blood count
- Musculoskeletal and connective tissue system: Tenosynovitis-like symptoms
- Vascular system: Hypotension, hypertension, deep vein thrombosis, pulmonary embolism, myocardial infarction, stroke, transient ischemic attack
- Respiratory system: Symptoms consistent with an asthmatic process
- Multi-system disorders: Symptoms consistent with fibromyalgia (e.g., joint and muscle pain, headaches, sleep disorders, gastrointestinal distress, and shortness of breath), individually and collectively.
- one prefilled dual-chamber syringe
- one plunger
- two alcohol swabs
- Advise females of reproductive potential of the possible risk to a fetus. Advise patients to inform healthcare provider of a known or suspected pregnancy
- If contraception is indicated, advise females of reproductive potential to use non-hormonal contraception during treatment with LUPRON DEPOT 3.75 mg
